Conference
Undiagnosed Day 2024
Undiagnosed Day 2024 350 million people live with an undiagnosed disease worldwide. Today we know that genome sequencing can provide answers …
Shinya Yamamoto, DVM, PhD. Department of Molecular and Human Genetics, Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital, Baylor College of Medicine, Houston, TX, USA
Agata Polizzi MD, PhD. National Centre for Rare Diseases, National Institute of Health, Rome, Italy
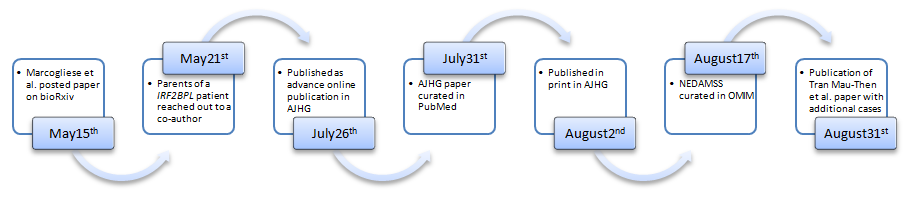
On May 21st this year, one of the authors of this article received an e-mail from the parents of a child that had been suffering from an undiagnosed neurological condition. The e-mail started out by describing the symptoms of their beloved son, a nine year old boy who was meeting his developmental milestones and was running around like any other child until his motor skills started to deteriorate around the age of three. Currently the patient is wheel chair bound and depends on G-tubes for his meals. The family had been seeking answers for their child’s conditions and have gone through their diagnostic odyssey for the past six years in the US as well as in Japan. They even have received three whole-exome sequencing from different laboratories, all of which came back with a negative result. One lab, however, noted a heterozygous truncating de novo variant in a gene called IRF2BPL, but the diagnostic lab could not tell the family what this meant since the gene was yet to be associated with a human disease. The reason they sent the e-mail was they found an article that I co-authored and was just posted on bioRxiv a week before on May 15th, titled “Loss-of-function in IRF2BPL is associated with neurological phenotypes”. The e-mail from the parents expressing their excitement that the paper helped them to finally find the answer the family was looking for years came as a big surprise. This was the first time we used a preprint server to disseminate our findings prior to the publication of the work in a peer-reviewed journal, and we had not imagined that someone outside of the biomedical science field will be paying attention to articles that are being posted in bioRxiv. This clearly meant that families of undiagnosed disease patients are really proactive in trying to get any information out on the internet that may help their loved ones, perhaps searching the name of the candidate disease gene ‘IRF2BPL’ on the internet on a regular basis to find out what a variant in this gene meant to their child.
bioRxiv is an online preprint server where any investigator can post scientific articles related to biology and medicine prior to its publication in peer-reviewed journals. Use of preprint servers such as arXiv has become a common routine in scientific disciplines such as mathematics and physics, fields in which peer-review processes can take a very long time. Through preprint servers, researchers can present their latest findings in a speedy manner, and readers can use their own judgement to interpret the manuscript. bioRxiv was launched in 2013 by the Cold Spring Harbor Laboratory to bring this custom into the biomedical research field. Although there are a number of concerns about such system where anyone can read papers that haven’t undergone a ‘quality control’ process offered by a peer-review process (1), a number of funding agencies including the NIH are beginning encourage the submission of preprints to speed up the dissemination of scientific and medical discoveries. Many scientific journals are now accepting submission of papers posted on preprint servers, and the IRF2BPL paper mentioned above was submitted to the American Journal of Human Genetics (AJHG) on the same day that was uploaded onto bioRxiv. The paper was successfully published online on July 26th after going through an authentic peer-review process for this journal. In this work, Marcogliese et al., report the identification and clinical evaluation of seven individuals with de novo nonsense, frame-shift or missense variants in IRF2BPL that exhibit overlapping neurological symptoms, a collaborative work by physicians and scientists of the Undiagnosed Diseases Network (UDN) and collaborators around the US and in Belgium (2). Interestingly, another paper from an independent group of clinicians and scientists from France, US, Netherlands, Switzerland and Russia was published in Genetics in Medicine on August 31st reporting 11 additional patients with de novo truncating variants in IRF2BPL (3). Most of the patients reported in the two papers show abnormal movement and gait, loss of speech, seizures, and many show developmental regression. Brain atrophy was also frequently seen in the patients in both papers, indicating a progressive neurodegenerative defect. Although the two papers propose different mechanisms of how the truncated IRF2BPL causes disease (Marcogliese et al., proposes a haploinsufficiency mechanism based on functional studies using Drosophila, whereas Tran Mau-Them et al., proposes a dominant negative or gain-of-function scenario), which need to be further explored through experimental studies in mammalian systems and by identifying additional patients, these papers define a new human disease entity for which at least 18 persons are known to be affected around the world.
On August 17th, a new disease entry was created on OMIM (Online Mendelian Inheritance in Man) under the name NEDAMSS (NEurodevelopmental Disorder with regression, Abnormal Movements, loss of Speech, and Seizures, MIM #618088) associated with IRF2BPL (MIM *611720). The curation of this new disorder in OMIM will likely lead to diagnosis of more individuals. Indeed, curation of a disease as an OMIM entry is an important milestone since this would lead to more clinical diagnostic laboratories tagging a specific gene in their clinical reports. By reviewing the chronological sequence related to the publication of Marcogliese et al. manuscript, we can see how, the use of a preprint server contributed to disseminate to a broader community, the information that pathogenic variants in IRF2BPL causes a neurological disease in humans, three months before the OMIM curation.
Of note, not all human genetics related papers in PubMed are captured in OMIM. The UDN for example have published several papers describing new human genotype-phenotype relationships prior to the publication of the IRF2BPL story that are yet to be curated in OMIM [ATP5F1D, Oláhová et al., AJHG (2/22/2018-online, 3/1/2018-print), TBX2: Liu et al., Human Molecular Genetics (5/2/2018-online, 6/15/2018-print), MTHFS; Rodan et al., Molecular Genetics and Metabolism (6/15/2018-online), TRAF7: Tokita et al., AJHG (6/28/2018-online, 7/5/2018-print)]. Interestingly, a couple of these studies were published prior to the IRF2BPL paper in the same journal (AJHG) so it is difficult to predict how some papers are prioritized for OMIM curation. According to the statistics, OMIM typically adds ~50 new disease entries and updates ~500 existing entries per month. This requires extensive efforts of curators with significant biomedical expertise at the McKusick-Nathans Institute of Genetic Medicine at the Johns Hopkins University School of Medicine to read, interpret and summarize the primary literature under the directorship of Dr. Ada Hamosh. It is possible that the explosion of new knowledge on human phenotype-genotype relationships based on whole-exome and whole-genome sequencing technology is over flooding their system to allow incorporation of all relevant literature into OMIM in a timely fashion. Also through personal communications with OMIM, we learned that OMIM only curate papers that have been officially printed and does not handle papers that are still at an ‘advanced online publication’ state. Therefore, even if there is a study that has been published in a peer-reviewed journal, OMIM search alone may miss some critical information that may aid in the diagnosis of an undiagnosed patient.
Keyword based searches on PubMed are complementary to fill the ‘OMIM gap’ but it is important to note that there is even a caveat here too. In some papers, just screening the title and abstract may not be sufficient to extract all of the contents that are published in the full manuscript (i.e. full spectrum of the phenotype and/or candidate gene). So if one does not read the entire paper, clinicians and human geneticists may not identify this information that maybe useful in clinical settings. In the future, integration of text-mining tools that can scan the full-text of papers that are available online may facilitate the identification of such articles that are missed through standard OMIM and PubMed searches. Finally, routine searches of preprint servers such as bioRxiv may facilitate the identification of papers that are still on the horizon. Although we would like to emphasize the caution one should pay attention to when reading non-peer reviewed manuscripts, such articles may provide some answers that undiagnosed patients and their family members have been waiting for years, just like in the example introduced in beginning of this story.
The families of the NEDAMSS patients are beginning to come together to exchange information and to further support basic, translational, and clinical research for this disease. Please visit the ‘IRF2BPL Support Group’ website at https://www.irf2bpl.org/ for more information.
[References Cited]
1. Sheldon T (2018) Preprints could promote confusion and distortion. Nature 559,445. PMID: 30042547
2. Marcogliese PC, Shashi V, Spillmann RC, Stong N, Rosenfeld JA, Koenig MK, Martínez-Agosto JA, Herzog M, Chen AH, Dickson PI, Lin HJ, Vera MU, Salamon N, Graham JM Jr, Ortiz D, Infante E, Steyaert W, Dermaut B, Poppe B, Chung H-L, Zuo Z, Lee P-T, Kanca O, Xia F, Yang Y, Smith EC, Jasien J, Kansagra S, Spiridigliozzi G, El-Dairi M, Lark R, Riley K, Koeberl DD, Golden-Grant K, Program for Undiagnosed Diseases (UD-PrOZA), Undiagnosed Diseases Network, Yamamoto S, Wangler MF, Mirzaa G, Hemelsoet D, Lee B, Nelson SF, Goldstein DB, Bellen HJ, Pena LDM (2018) Loss-of-function in IRF2BPL is associated with neurological phenotypes. AJHG. 103(2):245-260. PMID: 30057031, PMICD: 6081494
3. Tran Mau-Them F, Guibaud L, Duplomb L, Keren B, Lindstrom K, Marey I, Mochel F, van den Boogaard MJ, Oegema R, Nava C, Masurel A, Jouan T, Jansen FE, Au M, Chen AH, Cho M, Duffourd Y, Lozier E, Konovalov F, Sharkov A, Korostelev S, Urteaga B, Dickson P, Vera M, Martínez-Agosto JA, Begemann A, Zweier M, Schmitt-Mechelke T, Rauch A, Philippe C, van Gassen K, Nelson S, Graham JM Jr, Friedman J, Faivre L, Lin HJ, Thauvin-Robinet C, Vitobello A (2018) De novo truncating variants in the intronless IRF2BPL are responsible for developmental epileptic encephalopathy. Genet Med. 2018 Aug 31. doi: 10.1038/s41436-018-0143-0. [Epub ahead of print]. PMID: 30166628
[Key Websites]
bioRxiv : https://www.bioRxiv .org/
OMIM: https://www.OMIM.org/
PubMed : https://www.ncbi.nlm.nih.gov/PubMed/
Undiagnosed Diseases Network (UDN): https://undiagnosed.hms.harvard.edu/
IRF2BPL Support Group: https://www.irf2bpl.org/
Undiagnosed Day 2024 350 million people live with an undiagnosed disease worldwide. Today we know that genome sequencing can provide answers …
EURORDIS-Rare Diseases Europe is pleased to announce the 12th European Conference on Rare Diseases & Orphan Products (ECRD …
This conference is a unique platform that brings all parties involved in rare diseases to one place to obtain the most updated knowledge, increase …
13th Conference of Undiagnosed Diseases Network International 5-7 September, 2024, Seoul, Korea Overview UDNI 2024 Prof. Jong-Hee Chae is …